
All MS patients provided written informed consent for CSF sample collection and data analysis. This study was approved by the Ethics Committee of the University Hospitals Leuven (S60222 and S50354) and performed in accordance with the declaration of Helsinki. The use of post-mortem brain samples from the archives of the Center for Brain Research was approved by the Ethics Committee of the Medical University of Vienna (EK. Nr.: 535-2004).
Study design
In this study, we aimed to identify a strong predictive biomarker for disability progression in MS amongst five microglia/macrophage-related proteins measured in the CSF at diagnostic lumbar puncture – CHIT1, CHI3L1, sTREM2, GPNMB and CCL18. To this end, we extended our previously described cohort (n = 143)7 to 196 MS patients. Protein concentrations were measured in duplicate and blinded to clinical data. After rigorous quality control (QC), 192 MS patients were retained for further analyses. Primary and secondary endpoints were defined in advance. For single-time-point analysis, primary outcomes were ARMSS and annualized relapse rate (ARR) before treatment and secondary outcomes were MSSS and EDSS. For multi-time-point analysis (mixed-effects models and machine learning algorithms), we assessed EDSS as the only outcome. Next, we addressed the cellular source of CHIT1 – our most robust CSF biomarker – to provide a biological rationale for its prognostic value. We performed scRNA-seq on CSF of 11 MS patients and supplemented these data with single-cell profiles of MS CNS tissue from four publicly available sc/snRNA-seq datasets. Samples of interest were included based on prior knowledge as described below. All CSF and CNS transcriptomes that failed stringent QC were removed from downstream analyses. Finally, we corroborated our observed CHIT1 expression patterns at the protein level with immunohistochemistry on post-mortem brain tissue of MS patients, both in early and late disease stages. Tissue selection was informed by results from transcriptomics.
Biomarker analysis
Study population
All MS patients were diagnosed according to the 2017 revised McDonald criteria60 at the Department of Neurology of the University Hospitals Leuven (Belgium). We distinguished disease course based on a relapsing-remitting or primary progressive MS onset (RRMS or PPMS). Clinical data were collected by the same expert neurologist (BD) at diagnosis and follow-up visits up to 15 years (median of 5.4 years) after diagnosis, effectively minimizing information bias. All EDSS assessments were done when MS patients were not in active relapse. To control for the effect of age and disease duration on our disability measures, individual EDSS scores were converted to ARMSS and MSSS values, respectively61,62. The ARR was calculated over a minimum period of 90 days before the start of treatment, as reported earlier63. During disability follow-up (EDSS, ARMSS and MSSS), 62% of MS patients were on disease-modifying therapies.
CSF protein measurements
CSF of MS patients was collected during diagnostic lumbar puncture as part of standard clinical care. None of these patients were on disease-modifying therapy at the time of sample collection. Polypropylene tubes were centrifuged for 10 min at 3000× g at 4 °C and CSF supernatant was stored at −80 °C until protein measurements. We assessed CHIT1, CHI3L1, sTREM2, GPNMB and CCL18. NfL was also included as canonical biomarker for neuronal damage. CHIT1, GPNMB, CCL18 and NfL were measured within the entire MS cohort (n = 196). For CHI3L1 and sTREM2, measurements were limited to the original cohort (n = 143) as previously described7.
CSF protein concentrations were measured in duplicate and blinded to clinical data. For CHIT1, the CircuLex Human Chitotriosidase ELISA Kit (#CY-8074, MBL Life Science) was used. GPNMB and CCL18 concentrations were quantified with U-PLEX technology (#F21ZH-3 and #F212H-3, MSD). CHI3L1, sTREM2 and NfL measurements were obtained as previously described7. We performed all assays conforming to the manufacturer’s instructions.
QC was carried out as follows: we checked for consistency of the standard curves across plates, with 0.74 ˂ r ˂ 1 (P ≤ 0.05) for all assays. Concerning plate-to-plate variability, we calculated the coefficient of variation (CV) based on an internal control sample measured on all plates. Samples with a CV more than 20% or CSF biomarker concentrations below the detection limit were excluded. Samples above the upper detection limit were remeasured using a different dilution factor where possible or were eliminated otherwise. Within-plate variability was assessed based on the CV across duplicates (Supplementary Table 1). Mean values across duplicates were used for analysis.
Single-cell transcriptomics
Datasets
In-house CSF cohort: All patients (n = 11) were diagnosed with RRMS according to the 2017 revised McDonald criteria60 by the same expert neurologist (BD) at the Department of Neurology of the University Hospitals Leuven (Belgium) (Supplementary Data 2). None of these patients were on disease-modifying therapy at the time of sample collection.
Previously published CNS datasets: All previously published CNS datasets were downloaded as FASTQ files from the Sequence Read Archive [SRA, National Center for Biotechnology Information (NCBI)] under BioProject accession numbers PRJNA544731 [Schirmer et al.24], PRJNA749443 [Absinta et al.25], PRJNA743676 [Miedema et al.14] and PRJNA726991 [Trobisch et al.64]. The data from Miedema et al.14 was acquired via scRNA-seq, whereas the others performed snRNA-seq. From all studies, only samples of interest were used, i.e. CNS lesions or periplaque white matter of MS patients (Supplementary Data 2). CNS data was processed in parallel to our in-house CSF data from the Cell Ranger count pipeline onwards.
CSF sample collection and sequencing
CSF samples were obtained during diagnostic lumbar puncture. 5 ml of CSF was collected into round-bottom polypropylene tubes (Sarstedt) and processed within 1 hour after lumbar puncture to ensure optimal sample quality. CSF samples were centrifuged for 15 min at 400× g at 4 °C. We resuspended the CSF cell pellets in 100 µl of phosphate-buffered saline (PBS, Gibco) and counted the cells on a LUNA-FL Dual Fluorescence Cell Counter (Logos Biosystems). CSF cells were centrifuged again (15 min, 400× g, 4 °C) and resuspended in 1.5 ml of RPMI 1640 Medium (Gibco) containing 20% heat-inactivated fetal bovine serum (FBS, Merck) and 10% dimethyl sulfoxide (DMSO, Merck). CSF cells were transferred to Nunc CryoTubes (Thermo Scientific) and stored in a CoolCell container (Corning) at −80 °C. After 24 h, cells were relocated to liquid nitrogen for cryopreservation until further processing. Cryopreservation allowed us to limit batch effects without a substantial effect on gene expression patterns65,66.
Library preparation and scRNA-seq of our in-house CSF cohort was performed in three separate batches. The average latency between cryopreservation and library construction was 4.3 months (Supplementary Data 2). For every batch, we resuscitated the cryopreserved CSF cells in warmed RPMI 1640 Medium (Gibco) containing 20% heat-inactivated FBS (Merck). Cells were centrifuged for 10 min at 400× g at room temperature (RT) and resuspended in PBS (Gibco) containing 0.04% bovine serum albumin (BSA, Merck). We consistently obtained a high cell viability (mean: 94.7% ± 5.8% SD). Single-cell suspensions were loaded on a Chromium Next GEM Chip K for single-cell partitioning and barcoding with the Chromium Controller according to the Chromium Next GEM Single Cell 5′ Reagent Kit v2 (Dual Index) (#1000286 and #1000263, 10x Genomics). We performed further library preparation conforming to the manufacturer’s instructions. Libraries were sequenced on an Illumina NovaSeq 6000 with a target sequencing depth of 50,000 reads per cell.
Preprocessing of sequencing data
Processing of the sequencing data from both our in-house CSF cohort as well as the four previously published CNS datasets was performed with Cell Ranger v7.1.0 software (10x Genomics). Read alignment and transcript count were done individually for each sample via the Cell Ranger count pipeline with default parameters. Human reference GRCh38 (GENCODE v32/Ensembl 98) version 2020-A (July, 2020) was used for gene mapping.
Quality control
Before integration, all samples from all CSF and CNS datasets went separately through our QC pipeline in R v4.2.2. Ambient RNA contamination was removed using the SoupX v1.6.2 package67 with default parameters, except for samples from Miedema et al.14 where tfidfMin was adjusted to 0.8 (default 1) due to the lower cellular complexity of the dataset (prior FACS). Doublets were identified via scDblFinder v1.12.068 with default parameters. The corrected count matrices and relevant metadata were used to create Seurat objects for further analysis with Seurat v4.3.069. Features (genes) present in less than five cells were removed and only high-quality cells were retained, i.e. singlets with more than 200 features and a maximal percentage of mitochondrial RNA content of 15% for scRNA-seq data70 and 5% for snRNA-seq data25,64 (Supplementary Data 2). We normalized and scaled the count matrices via SCTransform v0.3.571 with default parameters. Mitochondrial and ribosomal content as well as the difference in expression between S and G2M cell cycle genes were regressed out.
Data integration, batch effect removal and myeloid clustering
In order to harmonize the different samples and avoid subject-specific clusters, we integrated the individual samples of the five datasets (our in-house CSF dataset and four previously published CNS datasets) into one dataset using the Harmony v0.1.1 package72 with default parameters. We assessed different integration strategies with various (combinations of) covariates to correct for. Best integration was obtained when each sample was treated as a batch and sequencing technique (scRNA-seq or snRNA-seq) was included as a variable. To cluster the cells, FindNeighbours() was run with Harmony-corrected principal components, followed by FindClusters(). We visualized the clusters on a Uniform Manifold Approximation and Projection (UMAP) plot. Optimal cluster resolution was determined with guidance of the clustree v0.5.0 package73. To identify myeloid clusters, we checked the expression of canonical myeloid marker genes (Supplementary Data 3). Furthermore, we used SingleR v2.0.074 for automated annotation based on the Novershtern immune reference dataset75 from the celldex v1.8.0 package74. Non-myeloid clusters were removed.
Annotation of myeloid clustering
DGE analysis was performed using FindAllmarkers() with default parameters, except for min.pct = 0.25 (default 0.10), to investigate the most differentially expressed genes (DEGs) in each cluster. We subdivided the myeloid clusters into monocytes, macrophages and microglia based on canonical marker genes (Supplementary Data 3). For more in-depth annotation of these clusters, we compiled gene sets of interest (or modules) from previously published articles (Supplementary Data 3) and scored their expression levels in our integrated dataset via AddModuleScore()76.
Pathway analysis
CHIT1+ cells were identified via WhichCells() as cells with an expression value > 0 in the RNA assay. We used FindMarkers() to get DEGs between CHIT1+ and CHIT1- cells. Only genes with an adjusted p-value < 0.05 were retained and used as input for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) gene set enrichment analysis via the clusterProfiler v4.6.2 package77.
Pseudotime trajectory analysis
To estimate cell state dynamics, we performed pseudotime trajectory analysis using the slingshot v2.6.078 and tradeSeq v1.12.079 packages. Homeostatic cluster “Microglia 1” (MG1) was defined as the starting point. Since the inclusion of non-microglia clusters [“Monocytes” (MON), “CNS-associated macrophages 1 and 2” (CAM1 and CAM2) and “Mixed myeloid cells” (MMC)] might lead to nonsensical trajectories, we subsetted our data to only retain microglia clusters for pseudotime trajectory analysis to gauge where the CHIT1+ microglia were positioned among the different microglia cell states. Moreover, we carried out DGE analysis along the inferred trajectories with fitGAM(), assoRes() and startVsEndTest().
Neuropathological evaluation
Patients
For neuropathological evaluation, formalin-fixed paraffin-embedded (FFPE) autopsy material from the archives of the Center for Brain Research (Medical University of Vienna) was used. We assessed samples from acute monophasic MS [n = 5, described before80], RRMS (n = 1), PPMS (n = 3) and SPMS (n = 3) patients. From these MS patients, we analyzed a total of 21 active (2 smoldering) and 8 inactive lesions (Supplementary Table 8).
Luxol Fast Blue-Periodic acid Schiff staining for myelin
5 µm thick paraffin sections of MS brain were stained with Luxol Fast Blue-Periodic acid Schiff (LFB-PAS) according to standard procedure to detect demyelinated lesions and macrophages with myelin degradation products.
Immune light microscopical staining
3-5 µm FFPE sections were deparaffinized in xylene (2× 15 min) and rinsed with 96% ethanol. To block endogenous peroxidase activity, samples were incubated in H2O2-methanol (30 min). Next, we gradually rehydrated the samples with ethanol (96%, 70%, 50%) and finally deionized water. Heat-induced epitope retrieval (HIER) was performed with ethylenediaminetetraacetic acid (EDTA, pH 9.0) in a household food steamer (Braun, 1 h). Afterwards, we rinsed the sections with tris-buffered saline solution (TBS) 3-5 times. Non-specific background reactions were blocked with 10% fetal calf serum (FCS) in DAKO buffer (Agilent) for 15 min. Hereafter, we incubated the slides with primary antibody CHIT1 (#HPA010575, Sigma, 1:100, 4 °C overnight) or CD68 (#M0814, Dakocytomation, 1:100, 4 °C overnight) and rinsed them with TBS before incubating them with a secondary biotin anti-rabbit-conjugated antibody (1:200, #711-065-152, Jackson, 1 h at RT) or biotin-anti-mouse-conjugated antibody (#715-065-150, Jackson, 1:500, 1 h at RT). Next, the slides were rinsed with TBS and incubated with peroxidase-conjugated streptavidin (1:500) in 10% FCS-DAKO buffer (1 h at RT). Finally, the sections were rinsed with TBS, developed with 3,3’-Diaminobenzidine (DAB) and counterstained in hematoxylin (15-20 s) before dehydration and mounting with cover slips using EUKITT (ORSAtec).
Immunofluorescent multiplex labeling
Immunofluorescent multiplex labeling was performed with Akoya’s Fluorescent Multiplex reagents according to the PerkinElmer´s application note ‘Automated multiplex Biomarker Staining and Imaging’ for CHIT1 (#HPA010575, Sigma, 1:1000), Iba1 (#019-19741, Wako, 1:10,000), PLP (#MCA839G, Bio-Rad, 1:5000), GFAP (#MS-1376, Thermo Scientific, 1:1000), TMEM119 (#HPA051870, Sigma, 1:1000) and CD68 (#M0814, Dakocytomation, 1:1000). In short, after deparaffination, the sections were fixed with 4% paraformaldehyde (PFA, 20 min), rinsed with deionized water and steamed in Antigen Retrieval buffer (pH 9.0, Akoya, AR900250ML) in a household food steamer (Braun, 1 h). The sections were rinsed with TBS-Tween 2.0 (TBST, pH 7.5) followed by a blocking step with Opal Antibody Diluent/Block solution (Akoya, ARD1001EA, 10 min). Next, we incubated the slides with a first primary antibody (4 °C, overnight). Then, the slides were rinsed with TBST and incubated with Akoya’s secondary antibodies (Opal Polymer horseradish peroxidase conjugated antibodies, ARH1001EA, 10 min at RT). The sections were rinsed with TBST and incubated with a fluorophore (Opal dye 480, 520, 570, 620, 690 or 780, 1:100, 10 min) in 1X Plus Amplification Diluent (Akoya, FP1497). Next, the slides were rinsed with TBST, fixed with 4% PFA (10 min) and steamed with Antigen Retrieval buffer (AR6, Akoya, AR600250ML, 30 min). From here, the above-mentioned steps were repeated for each primary antibody. Finally, we counterstained with 4’,6-Diamidino-2-phenylindole (DAPI) and the slides were mounted with Fluoromount-G (SouthernBiotech). The slides were scanned with a Vectra Polaris scanner (PerkinElmer) at 20× magnification or on a laser scanning microscope equipped with lasers for 504, 488, 543 and 633 nm excitation (Leica SP5, Leica, Mannheim, Germany).
Statistical analysis
For single-time-point analysis and mixed-effects models, all statistical tests were performed with R v4.0.1. Normal distribution of the data was assessed with Shapiro-Wilk tests. Biomarker concentrations and clinical variables were normally distributed after a logarithmic transformation with base 10 and an inverse rank normal transformation, respectively.
Single-time-point analysis
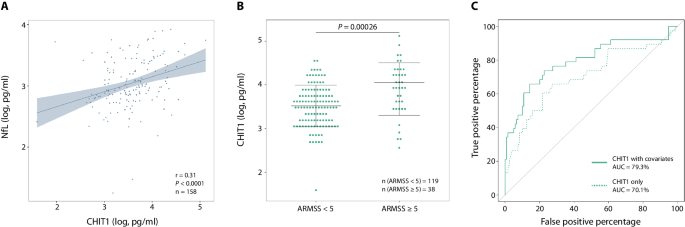
To evaluate the relationships between the five microglia/macrophage CSF biomarkers – CHIT1, CHI3L1, sTREM2, GPNMB and CCL18 – and CSF NfL concentrations, we calculated pairwise Pearson correlation coefficients and tested their significance with two-sided Pearson correlation tests. Next, we tested the association between these biomarker concentrations and several MS disease activity parameters: ARMSS and ARR before treatment (primary outcomes); MSSS and EDSS (secondary outcomes). These associations were assessed in a linear regression model which included age at diagnosis and sex as covariates as well as the rs150192398 genotype for CHIT1 as in our previous work7. Based on the results of the linear regression model, we focused on the correlations between CSF CHIT1 concentrations and future disability (ARMSS, MSSS and EDSS). The percentage of variance explained (r2) with and without clinical covariates, the division between MS patients with high (ARMSS ≥ 5) and low (ARMSS < 5) disability accumulation as well as the ROC curve were determined as previously described7,61. We applied Bonferroni corrections for multiple testing as stated in the table legends.
Multi-time-point analysis – mixed-effects models
The correlation between CSF CHIT1 concentrations at diagnosis and disability progression (EDSS) over time was assessed using both mixed-effects models and artificial intelligence algorithms (see Machine learning algorithms). Mixed-effects models were chosen due to their robustness and power to handle unbalanced data given the gradual loss of EDSS data over time within our MS cohort. Despite the unbalanced EDSS data, the mixed-effects models allowed us to examine the impact of CHIT1 on disability progression over time while also considering variability within and across MS patients and time as well as controlling for relevant variables81. Within our model development, we tested models with and without the following considerations: controlling for 1) interindividual variance in EDSS scores; 2) intraindividual variance in EDSS scores across time; and 3) interaction effects between CHIT1 and time from MS diagnosis to EDSS score. Mixed-effects models were constructed and assessed with packages lme4 v1.1-31, afex v1.2-1 (convergence checks) and ggplot v3.4.2. Included continuous predictors were scaled to assist with convergence and result interpretation. Model fits were assessed using likelihood-ratio tests.
Multi-time-point analysis – machine learning algorithms
In D’hondt et al.19, we developed machine learning algorithms to model EDSS trajectories of MS patients. In short, this approach transformed real-world EDSS assessments for each patient to yearly average disability scores up to the 10th year after diagnosis. These 10 disability averages then formed the outcomes of a machine learning model based on random forests. For each of these 10 outcomes, a random forest was trained. In a regressor chain fashion82, the disability predictions of previous years were added as extra input to the random forests of the next years. These trained models could then be used to predict the disability of an individual MS patient for up to 10 years after diagnosis.
In this work, we used this regressor chain model to analyze the prognostic importance of input variables, in particular the CSF biomarkers under study. For this, we used permutation importance scores20, which reflect the effect on model performance if one of the input variables is randomly shuffled (simulating the removal of that variable). Model performance (mean absolute error and Pearson correlation) was measured using 5-fold cross-validation (to avoid overfitting) and the variable shuffling was repeated multiple times to counter variability. In order to control for statistical interactions between CSF biomarkers, we conducted a separate analysis where we made a correction on the permutation importance scores for possible CSF biomarker interactions. To this end, in addition to shuffling each biomarker separately, we also shuffled all possible pairs, triplets, quartets and quintets of biomarkers (simulating the removal of several biomarkers simultaneously). In each of these shuffles, the performance drop was calculated and divided by the size of the shuffled subset of biomarkers (1, 2, 3, 4 or 5) and by the number of times each biomarker appears in a subset of that size (1, 5, 15, 5, 1). For each biomarker, a final score was then obtained by summing all performance drops in which that biomarker was shuffled. The machine learning methodology was developed in Python 3.8.10.
Single-cell transcriptomics
With regard to the transcriptomics, all statistical tests were performed in R v4.2.2. DGE results were consistently assessed for statistical significance using a two-sided unpaired Wilcoxon rank-sum test with Bonferroni correction for multiple testing. Differential expression of modules across clusters was demonstrated using a Kruskal-Wallis test followed by a two-sided Dunn’s test for pairwise multiple comparisons with Benjamini-Hochberg correction for multiple testing. For pathway analysis, p-values were calculated by a one-sided Fisher’s exact test with Benjamini-Hochberg correction. For the DGE analyses across trajectories, statistical significance was evaluated by means of a two-sided Wald test with Benjamini-Hochberg correction for multiple testing. The exact method is also always stated in the legends. For histology, no statistical analyses were performed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.